
科技课堂:Parallel-META 3:综合自动分析流程和使用说明
Parallel-META 3是由中国科学院青岛生物能源与过程研究所单细胞研究中心开发的综合的自动分析流程软件包。可以用来分析宏基因组、16S/18S rRNA测序数据。通过并行算法和优化,软件与其他传统主流流程相比实现了快速自动分析。
http://bioinfo.single-cell.cn/parallel-meta.html
Single-cell Reseearch Center of Qingdao Institute of BioEnergy and Bioprocess Technology, Chinese Academy of Sciences (QIBEBT-CAS, 中国科学院青岛生物能源与过程研究所).
1. Jing., et al., Parallel-META 3: Comprehensive taxonomical and functional analysis platform for efficient comparison of microbial communities, Scientific Reports, 2017.
2. Su X., Pan W., et al.,Parallel-META 2.0: Enhanced Metagenomic Data Analysis with Functional Annotation, High Performance Computing and Advanced Visualization, PLoS ONE, 2014.
3. Su X., et al. Parallel-META: Efficient Metagenomic Data Analysis Based on High-Performance Computation, BMC Systems Biology, 2012.
下载地址:
http://bioinfo.single-cell.cn/parallel-meta.html
软件拥有Linux(32位和64位)和Mac OS X版本。
软件依赖:
R3.0或以上(Linux自带);Bowtie2 (2.1.0或更高版本, 已集成在安装包中);HMMER 3(3.0或更高版本, 已集成在安装包中);Compiler 源码安装时要求(Linux需要g++4.0或以上版本,Mac OS X需要g++-6 6.0或以上版本)
虽然有依赖,但软件本身提供的一行命令会自动安装相关依赖。
软件安装:
解压:tar –xzvf parallel-meta-3.tar.gz
安装:cd parallel-meta
./install.sh
注意事项:
软件输出的文件会默认覆盖原文件夹的内容,输出时注意文件位置。
1 文件输入格式:
Parallel-META 3有自己的文件输入格式。
(1)实验设计文件:(Meta-data format)

实验设计文件中每一行代表一个样品,每一列代表一个特征。首格样品名称不能以#或者数字起始;且样品名不能有空格(‘ ’),反斜线('/'),或者tab建(‘\t’)。特征不能为‘NA’或‘na’,可以使用‘Unspecified’等。
(2)样品列表文件:(Sequence format and sequence list)
Parallel-META 3通常接受拆分的序列,格式为一个样品一个Fasta/Fastq文件。序列标签不能包含
文件可以放在一个文件夹下,每一条序列标签名称不能有空格(‘ ’),反斜线('/'),或者tab建(‘\t’),每一条序列为一行。16S rRNA序列,软件支持 -R 选项输入双端序列;宏基因组只支持单端测序文件。
例:PM-pipeline流程,单端序列列表可以如下:
/home/data/sample1.fasta
/home/data/sample2.fasta
/home/data/sample3.fasta
(将sample可替换为自己的编号)
双端序列列表:
/home/data/sample1_pair1.fasta
/home/data/sample1_pair2.fasta
/home/data/sample2_pair1.fasta
/home/data/sample2_pair2.fasta
/home/data/sample3_pair1.fasta
/home/data/sample3_pair2.fasta
/home/data/sample4_pair1.fasta
/home/data/sample4_pair2.fasta
(将sample,pair可替换为自己的编号)
对于全部样品放在一个文件中的情况,软件提供了PM-split-seq命令,可以将样品按照barcode或者分组信息拆分为可用于Parallel-META 3流程的文件列表
PM-split-seq
首先介绍样品拆分命令。支持fastq和fasta文件。因为软件支持的是 单文件夹+文件夹中文件列表 的输入形式。
PM-split-seq -i seq.fa -b barcode.txt -o seq.out
命令支持以barcode拆分序列。barcode.txt文件格式:
ATTCGT Sample1
AGCGTC Sample2
……..
CGTGAC SampleN
PM-split-seq -i seq.fa -g seq.groups -o seq.out
命令支持以分组信息拆分序列。seq.groups文件格式(Mothur格式):
Seq_Id_1 Sample1
Seq_Id_2 Sample2
……
Seq_Id_N SampleN
PM-split-seq -i seq.fa -q T -o seq.out
-q T参数可以以QIIME格式拆分文件。
QIIME命名格式为:
groupname1_1
groupname1_2
……
groupname2_1
groupname2_2
……
PM-pipeline
全自动分析流程。
PM-pipeline -D B -m design.txt -i sammple.list -o result/
-D 选择序列文件属性,B(16S rRNA),E(18S rRNA),默认B
-m 实验设计文件(1)
-i 样品列表文件(2)
-o 输出文件夹
一般可选参数选择默认即可。
可选参数:
-M 序列属性,T(宏基因组序列)或F(rRNA序列),默认F
-e 比对模型 0:非常快,1:块,2:敏感,3非常敏感,默认3(此处应该是比对的准确性和效率之间的调整,牺牲准确性会带来高速的比对)
-P 双端序列方向,0:正&反,1:正&正,2:反&正,默认0
-r RNA拷贝数校准,T或F,默认T
-a rRNA提取的长度临界值,0是不提取,默认0(有-M T参数时使用)
-k 序列格式检查,T或F,默认F
-f 功能分析,T或F,默认T
统计参数:
-L 分类级别(1~6:门~种),多级别输入支持
-W 分类学距离种类:0:weighted,1:unweighted,2:both。默认2
-F 功能级别(Level 1,2,3,4(KO级别)),多级别输入支持
-S 序列标准化,默认为0,填数字N为随机抽取N条序列
-B Bootstrap验证次数,默认200 最大值1000
-R 稀释曲线,T或F,默认F
-E 序列是否是成对的,T或F,默认F(也没懂)
-C Cluter number, default is 2(没懂,这个是啥)
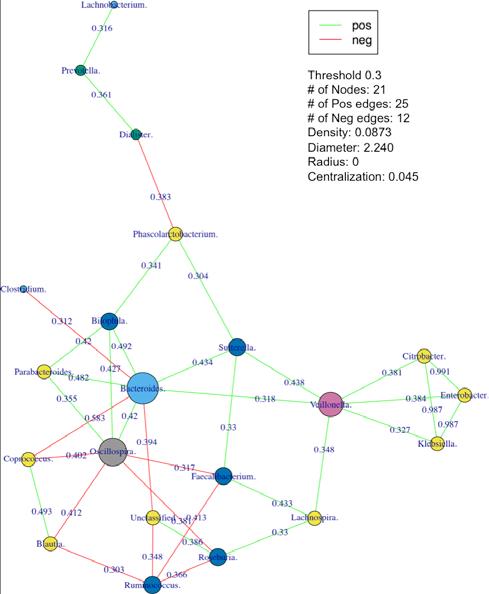
-T Network分析边缘的临界值,默认0.5,应该是0.5以上的才计入统计绘图。
其他选项参数:
-t cpu核数,默认自动
-h 帮助
注意:
1 实验设计文件序列文件顺序(特别是双端文件)要和样品列表文件一致
2 稀释曲线默认不计算,使用-R T参数调整
3 序列抽平标准化默认不做,使用-S来设置抽平深度
其余的命令参数参考http://bioinfo.single-cell.cn/parallel-meta.html中的手册。这里简单的介绍一下其他命令。
PM-parallel-meta:单端测序序列分析。主要做宏基因组分析的自动流程。
PM-parallel-meta –m meta.fasta –o metaresults –L 150
-L rRNA抽出的阈值,默认0
PM-format-seq:检查序列格式,并生成Parallel-META 3可识别的格式。这个命令很重要,可以检查并标准化自身文件的序列名称。官方也建议先做这一步。
PM-format-seq –i sample.fasta
或
PM-format-seq –l list.txt
PM-update-taxa:将分类和注释信息的格式升级到最新版本。list.txt文件在注释分析结果文件夹(*.List/)。这个是针对2.0老版本的“补丁”,官方建议重新用新流程再分析一遍。
PM-update-taxa –l list.txt
PM-extract-rna(包含在PM-parallel-meta中):从宏基因组测序结果中提取rRNA
PM-extract-rna –m examples/meta.fasta –o metaresults –e 1e-20 –l 150
PM-class-tax(包含在PM-parallel-meta中):序列注释分析。可输入单端测序文件和文件列表,或者OTU table (Abundance_Tables)
PM-class-tax –l list.txt –o result_plot
或
PM-class-tax –T Sample.OTU.Count –o result_plot
PM-class-func(包含在PM-parallel-meta中):序列功能分析。可输入单端测序文件和文件列表,或者OTU Abundance_Tables
PM-class-func –l list.txt –o result_func
或
PM-class-func –T Sample.OTU.Count –o result_func
PM-class-func-nsti(包含在PM-parallel-meta中):利用NSTI值 (Nearest Sequenced Taxon Index)估算序列功能。可输入单端测序文件和文件列表,或者OTU Abundance_Tables
PM-class-func-nsti –l list.txt –o result_func
或
PM-class-func-nsti –l Sample.OTU.Count –o result_func
PM-taxa-sel:从多序列中按注释分类层级提取序列。
PM-taxa-sel –l list.txt –o taxa.txt –L 6
或
PM-taxa-sel –T Sample.OTU.Count –o taxa.txt –L 6
PM-func-sel:从多序列中按KEGG通路分类层级提取序列。
PM-func-sel –l list.txt –o func.txt –L 2
或
PM-func-sel –T Sample.KO.Count –o func.txt –L 2
PM-comp-sam:基于注释信息用于多序列比较和相似性计算。
PM-comp-sam –l list.txt –o sim_matrix.txt –t 8
或
PM-comp-sam –T Sample.OTU.Count –o sim_matrix –t 8
PM-comp-sam-func:基于功能注释用于多序列比较和相似性计算。
PM-comp-sam-func –l list.txt –o sim_matrix.txt –t 8
或
PM-comp-sam-func –T Sample.KO.Count –o sim_matrix.txt –t 8
PM-rare-curv:用于稀释曲线的分析和作图。
PM-rare-curv –i taxa.Count –o rare-out –b 20
PM-comp-corr:用于物种注释和功能注释的相关计算。支持PM-taxa-sel和PM-func-sel的输出结果。
-f 0:(Spearman) 或1:(Pearson) 算法,默认 0
-T 网络分析阈值, 默认0.7
PM-comp-corr –i taxa.txt –o taxa.network.txt
PM-split-table:根据相对丰度表和绝对数量表,以实验设计文件为参照拆分序列文件。支持PM-taxa-sel和PM-func-sel的输出结果。
PM-split-table –i taxa.txt –m meta.txt –g Status –o taxa.groups
PM-split-matrix:用于拆分相似距离矩阵。支持PM-taxa-sel和PM-func-sel的输出结果。
PM-split-matrix –i matrix.txt –m meta.txt –g Status –o matrix.groups
R环境相关命令:
PM_Config.R:用于安装软件需要的R依赖。
Rscript PM_Config.R
PM_Distribution.R:用于画物种注释和功能注释的堆积柱状图。集成在PM-taxa-sel和PM-func-sel中,参数为-p,也可对其生成的结果作图。
PM_Heatmap.R:画heatmap图,集成在PM-comp-sam和PM-comp-sam-func中,参数为-p,也可对其生成的结果作图。
PM_Hcluster.R:分级聚群图。集成在PM-comp-sam和PM-comp-sam-func中,参数为-p,也可对其生成的结果作图。
PM_Pcoa.R:PCoA作图。可对PM-comp-sam和PM-comp-sam-func生成的结果作图。
PM_Pca.R:PCA作图。可对PM-taxa-sel和PM-func-sel生成的结果作图。
PM_Adiversity.R:α多样性作图。可对PM-taxa-sel和PM-func-sel生成的结果作图。
PM_Bdiversity.R:β多样性作图。可对PM-comp-sam和PM-comp-sam-func生成的结果作图。
PM_Marker_Test.R:通过离散变量(discrete variables)寻找Biomarker。可对PM-taxa-sel和PM-func-sel生成的结果作图。
PM_Marker_RFscore.R:利用随机森林算法为Biomarker赋分。可对PM-taxa-sel和PM-func-sel生成的结果作图。
PM_Marker_Corr.R:通过连续变量(continuous variables)寻找Biomarker。可对PM-taxa-sel和PM-func-sel生成的结果作图。
PM_Network.R:代谢共网络分析。功能集成在PM-comp-corr中,参数-N。可对PM-comp-corr和PM-comp-sam和PM-comp-sam-func生成的结果作图。
用PM-pipeline分析后会生成一系列文件/文件夹,命名为“taxa”为物种注释结果,“func”为代谢功能预测结果。
index.html
可通过网页超链接的形式查看结果。

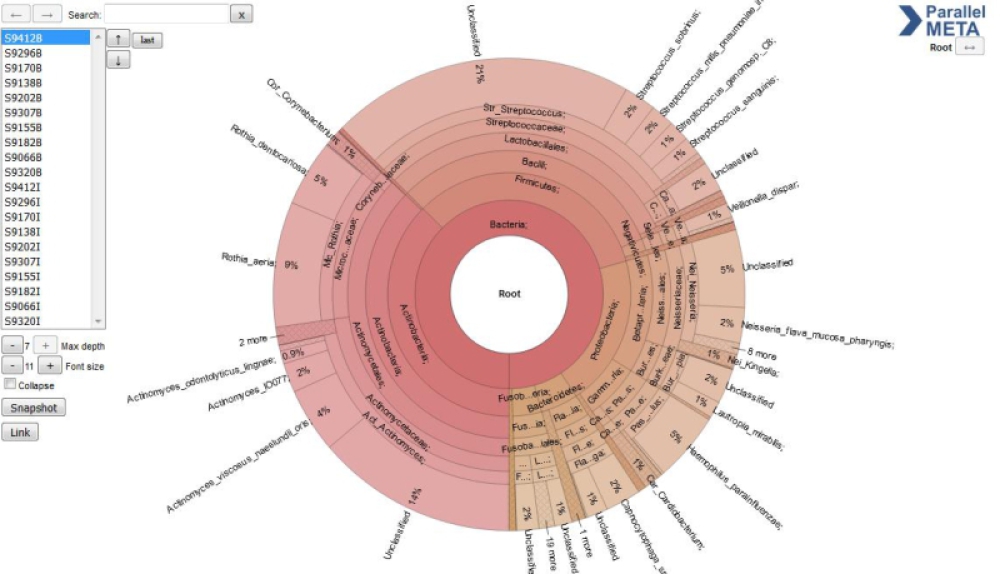
Sample_Views
包含每个样本的交互式饼图
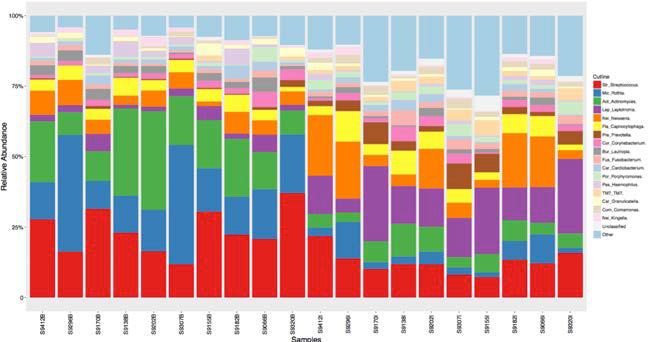
Abundance_Tables
包含相对丰度表(*.Abd),OTU绝对序列计数表(*.Count),柱状图(*Abd.pdf)。包含不同的分类层级。
Distance_Matrix
距离矩阵。包含所有样品成对的距离矩阵(*.dist),基于OTUs和KO的无监督聚类结果(*.dist.clusters.pdf and *.dist.heatmap.pdf)距离基于Meta-storms 算法。Meta-storms algorithm (Su, et al., Bioinformatics, 2012, Figure 4).

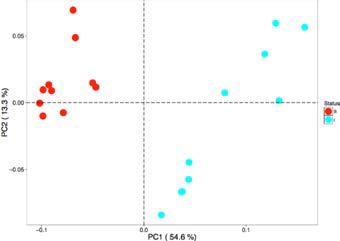
Clustering
聚类。包括PCA和PCoA结果。
Alpha_Diversity
α多样性结果图和统计分析结果(*.Alpha_diversity_Boxplot.pdf and *.Alpha_diversity_Index.txt)有参数时生成稀释曲线。p值为秩和检验rank-sum tests。

Beta_Diversity
β多样性。包含多元统计分析结果(*.Beta_diversity_Boxplot.pdf and *.taxa.dist.Beta_diversity_Values.txt),p值用的是Adonis/Permanova tests

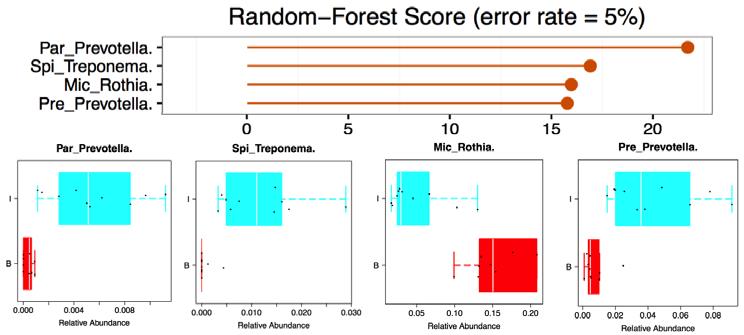
Markers
包含不同组间的Biomarker物种((*.sig.boxplot.pdf and *.sig.meanTests.xls)和随机森林算法分值((*.RFimportance.pdf and *. RFimportance.txt)
Network
网络图。(*.network.pdf)基于不同物种和功能注释。
本流程已经过测试,正式上线我所微生物组生信平台。
附件下载: